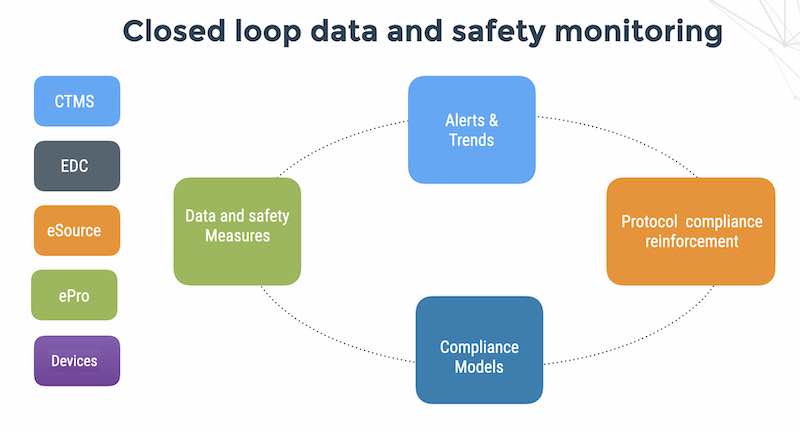
At a site monitoring visit, the CRA works on assuring protocol compliance. Assuring protocol compliance is one of the 3 pillars of GCP:
The 3 pillars of GCP (good clinical practice)
1. Patient safety
2. Protocol compliance
3. Data quality
(We note that setting the focus on the primary clinical and safety end-points results in formulation of GCP as an exercise in optimizing patient compliance to the protocol.)
With the understanding that clinical trial site monitors commonly use checklists for their site visits our first question is to challenge the utility of checklists:
To what extent do fixed checklists enable the study monitor and sponsor
to assess the impact that study deviations have on protocol compliance?
Take for example the activity of monitoring IC (informed consent); a best practice informed
consent monitoring checklist looka like this:
Informed consent monitoring checklist
1. Was the consent form used, and translated versions, approved by the IRB?
2. Was the ICF the most current and approved version?
3. If the consent is available in more than one language, was the participant given a chance to choose the language he/she prefers?
4. Did the participant receive full explanation of the contents of the ICF?
5. Did the participant have ample time to ask any questions and were they addressed adequately? Was the ICF signed before any study procedures? (N/A if the trial has received an exemption from IRB to consent after some study procedures)
6. If the subject is unable to read, was an independent witness present throughout the consent process?
7. Was the participant coerced?
8. Did the participant apparently understand the contents of the ICF?
9. Was IC form signed appropriately?
10. Was the environment suitable for the IC process?
(Courtesy of Global Health Trials)
You can check-off 11 items on the list but there is only 1 question that matters:
“Are there patients participating now in the study who did not sign the ICF (informed consent form)”
Why does the version or the environment matter if the patient is enrolled without informed consent? How does this checklist evaluate the impact of deviations? Does the checklist provide any quantitative measures of patient compliance?
After you ask that 1 question – (Are are any patients enrolled who did not sign ICF?) you can go on to quantify the impact (by asking how many patients are enrolled in the study without signed ICF) and then proceed to provide corrective and preventive actions.
In this article we suggest considering an alternative approach based on generating and analyzing multiple threat scenarios for the clinical study being monitored.
Since clinical trial data is highly-dimensional (typically 500-1000+ dimensions) we may reap significant benefits from this approach since with so many dimensions there tend to be many unconnected and undiscovered stovepipes of compliance and data governance.
Multiple threat scenarios enable auditors and study monitors to side-step large scale self-assessment checklists and problematic integration of data across stovepipes (large drug studies and large CROS like Quintiles, PPD and ICON typically use multiple systems from multiple vendors creating multiple unconnected stovepipes of data – one of the key reasons it takes 5-7 weeks to respond to a deviation) and focus on key assets, attacks and common vulnerabilities in key operational processes of the clinical trial like informed consent, eligibility criteria and treatment compliance (whether treatment is self-administered by the patient or administered by medical staff in a hospital).
In our experience, the sponsor is primarily interested in how cheaply the audit can be done and how much time and money they can save further down the road. For the business unit developing the medical device or drug, using a technique of multiple threat analysis will help show the best and most cost-effective way to progress from audit to patient compliance.
Do you base your regulatory affairs policy on Google?
You can do some homework online and then hire a clinical regulatory and compliance consultant who will walk you through the various GCP requirements and help you implement as many items as possible. This seems like a reasonable approach, but the more controls you implement, the more money you spend and moreover, you do not necessarily know if your risk posture has improved since you have not examined your value at risk – i.e how much money it will cost you for rework if more patients have to be enrolled due to non-adherence to protocol. Recall that patient protocol compliance is central to the success of your clinical trial and the defense of your claims with the FDA should rely on your experimental design, data and risk-analysis and not on the percentage SDV (source document verification) that study monitors performed.
Top-down risk-analysis
Taking a page out of the privacy and security playbook, we want to do a top-down risk analysis, and then continue with risk management and periodic protocol compliance activity review during the course of the clinical trial.
The best way to do that top down risk analysis is to build probable threat scenarios – considering what could go wrong – sites doing shoddy data entry or a hacker sniffing the hospital wired LAN for PHI and destroying the integrity of your randomized controlled trial.
Threat scenarios as an alternative to compliance check lists
When we perform a software security assessment of a medical device or healthcare system, we think in terms of “threat scenarios” or “attack scenarios”, and the result of that thinking manifests itself in planning, penetration testing, security countermeasures, and follow-up for compliance. The threat scenarios are not “one size fits all”.
The threat scenarios for clinical trials for AIDS diagnostics using medical devices that automatically scan and analyze blood samples, or an Army hospital using a networked brain scanning device to diagnose soldiers with head injuries, or an implanted cardiac device with mobile connectivity or immunotherapy treatment for cancer are all totally different.
We evaluate the medical device / investigational product from an attacker point of view, then from the management team point of view, and then recommend specific cost-effective, security countermeasures to mitigate the damage from the most likely attacks.
In our experience, building a risk control portfolio based on attack scenarios has 3 clear benefits;
1. A robust, cost-effective monitoring portfolio based on attack analysis results in robust compliance over time since you now have a formal methodology for evaluating new emerging issues such as mobile devices or changes to regulation.
2. Executives related well to the concepts of threat modeling / attack analysis. Competing, understanding the value of their assets, taking risks and protecting themselves from attackers is really, at the end of the day why executives get the big bucks.
3. Threat scenarios are a common language between IT, clinical operations teams and the business area managers. This last benefit is extremely important in your organization, since business delegates compliance to regulatory affairs and regulatory affairs delegates assessment to the site monitor teams and there is clearly a disconnect by the time you go from a business manager to a CRA.
As I wrote in a previous essay “The valley of death between IT and security“, there is a fundamental disconnect between IT operations (built on maintaining predictable business processes) and security operations (built on mitigating vulnerabilities).
The disconnect between sponsor business management and site monitors.
Business executives delegate clinical operations to VP Clinical who delegates to CROs who delegate compliance to sites on the tacit assumption that each are the experts in their own particular domain. This is a necessary but not sufficient condition.
In the current environment of rapidly evolving types of attacks (hacktivisim, nation-state attacks, credit card attacks mounted by organized crime, script kiddies, competitors and malicious insiders and more…), it is essential that business managers, sites and regulatory affairs professionals, communicate effectively regarding the types of attacks that their organization may face and what is the potential business impact on the clinical trial.
If you have any doubt about the importance of sponsors sharing data with sites, consider that leading up to 9/11, the CIA had intelligence on Al Qaeda terrorists and the FBI investigated people taking flying lessons, but no one asked the question why Arabs were learning to fly planes but not land them.
With fundamental disconnects between 3 key stakeholders of clinical data (sites, monitors and sponsors), it is no wonder that organizations are having difficult assessing GCP compliance in a timely fashion –
Sponsors, monitors and sites (and increasingly patients) need a common language to execute their mission, and I submit that building risk control portfolio de your clinical trial around most likely threat scenarios from an attacker perspective is the best way to cross that valley of death.
There seems to be a tacit assumption with pharma and medtech executives that regulatory compliance is already a common language of compliance for a clinical trial, but as we demonstrated at the beginning of this article, compliance checklists like ICF monitoring etc, are a dangerous replacement for not thinking through the most likely threats to your clinical trials.
Let me illustrate why compliance checklists are not the common language we need by taking an example from another compliance area – credit cards.
PCI DSS 2.0 has an obsessive preoccupation with anti-virus. It does not matter if you have a 16 quad-core Linux database server that is not attached the Internet with no removable device nor Windows connectivity.
PCI DSS 2.0 wants you to install an anti-virus and open the server up to the Internet for the daily anti-virus signature updates. This is an example of a compliance control policy that is not rooted in a probable threat scenario that creates additional vulnerabilities for the business.
Consider some deeper ramifications of check-list-based compliance to the protocol.
When a QSA or HIPAA auditor records an encounter with a customer, he records the planning, penetration testing, controls, and follow-up, not under a threat scenario, but under a control item (like access control). The next auditor that reviews the compliance posture of the business needs to read about the planning, testing, controls, and follow-up and then reverse-engineer the process to arrive at which threats are exploiting which vulnerabilities.
In the cyber security space, actors such as government agencies (DHS for example) and security researchers go through the same process. They all have their own methods of churning through the planning, test results, controls, and follow-up, to reverse-engineer the data in order to arrive at which threats are exploiting which vulnerabilities.
This ongoing process of “reverse-engineering” is the root cause for a series of additional problems:
1. Lack of overview of the the threats and vulnerabilities to clinical trials that really count.
2. No sufficient connection to best practice controls, no indication on which controls to follow or which have been followed.
3. No connection between controls and protocol deviation events, except circumstantial.
4. No ability to detect and warn for negative interactions between controls (for example – edit checks that generate large number of queries on every field, hobbling the ability of the sites to collect data in a timely manner).
5. No archiving or demoting of less important and solved threat scenarios (since the checklists are control based).
6. Lack of overview of compliance status of a particular site, only a series of historical observations disclosed or not disclosed. (Is Bank of America getting better at data security or worse? Is the Department of Clinical Neuropathology at King’s College Hospital getting better at GCP compliance or worse?)
7. An excess of paper documents that cannot possibly be read by the regulatory and clinical affairs manager at every encounter.
8. Regulatory and data borders are hard to define since the border definitions are networks, systems and applications not
Beyond checklists – using value at risk to assess impact of patient compliance violations
Checklists are good for ensuring a repeatable process but threats to your study are rooted in unforeseen events like patients without informed consent. Your threat scenarios should consider your study assets (your data, systems, management attention, reputation) values, vulnerabilities, threats and effective security countermeasures.
Threat analysis as a methodology for monitoring your clinical trial does not count activities like site visits and SDV. It is a systematic way to help you consider the fastest and most cost-effective way to reduce your risks of protocol non-compliance, safety and data quality.